From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis
 Marius van den Beek
Marius van den Beek
 Dave Clements
Dave Clements Daniel Blankenberg
Daniel Blankenberg Anton Nekrutenko
Anton NekrutenkoOverviewQuestions:
Objectives:
Learn how to get and use data from the Sequence Read Archive in Galaxy.
Requirements:
Understand how Galaxy and the Sequence Read Archive interact.
Be able to go from Galaxy to the Short Reach Archive, query SRA, use the SRA Run Selector to send selected metadata to Galaxy, and then import sequence data from SRA into Galaxy.
- Introduction to Galaxy Analyses
- Sequence analysis
- Quality Control: slides slides - tutorial hands-on
- Mapping: slides slides - tutorial hands-on
Time estimation: 45 minutesSupporting Materials:Last modification: May 18, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITShort Link: https://gxy.io/GTN:T00315
Introduction
The aim of this tutorial is to introduce you to the processing of next generation sequencing data in Galaxy. This tutorial uses a COVID-19 variant calling from Illumina data, but it isn’t about variant calling per se.
At the completion of this tutorial you will know:
- How to find data in SRA and transfer this information to Galaxy
- How to perform basic NGS data processing in Galaxy including:
- Quality Control (QC) of Illumina data
- Mapping
- Removal of duplicates
- Variant calling with
lofreq - Variant annotation
- Using datasets collections
Find necessary data in SRA
First we need to find a good dataset to play with. The Sequence Read Archive (SRA) is the primary archive of unassembled reads operated by the US National Institutes of Health (NIH). SRA is a great place to get the sequencing data that underlie publications and studies. Let’s do that:
Hands-on: Task description
- Go to NCBI’s SRA page by pointing your browser to https://www.ncbi.nlm.nih.gov/sra
- In the search box enter
SARS-CoV-2 Patient Sequencing From Partners / MGH:(Alternatively, you simply access the data via link)
- Note that some of the datasets found say “ARTICv3 amplicon sequencing”. This is a sequencing technique that requires addition analysis steps not discussed in this tutorial. The data that we will analyse (datasets mentioned below) uses a technique called “metagenomic sequencing”.
- The web page will show a large number of SRA datasets (at the time of writing there were 3,927). This is data from a study describing analysis of SARS-CoV-2 in Boston area.
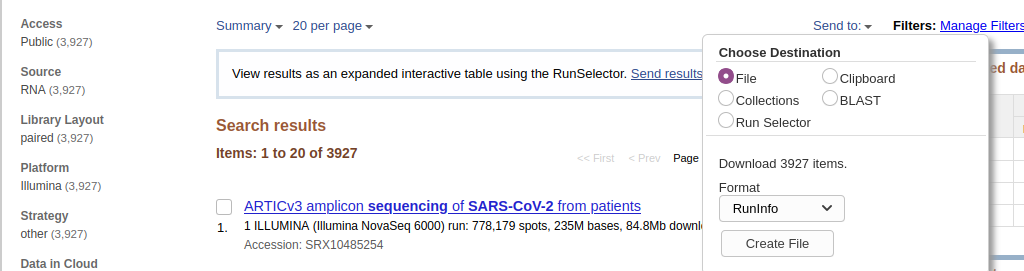
- Download metadata describing these datasets by:
- clicking on Send to: dropdown
- Selecting
File- Changing Format to
RunInfo- Clicking Create file Here is how it should look like:
- This would create a rather large
SraRunInfo.csvfile in yourDownloadsfolder.
Now that we have downloaded this file we can go to a Galaxy instance and start processing it.
CommentNote that the file we just downloaded is not sequencing data itself. Rather, it is metadata describing properties of sequencing reads. We will filter this list down to just a few accessions that will be used in the remainder of this tutorial.
Process and filter SraRunInfo.csv file in Galaxy
Hands-on: Upload `SraRunInfo.csv` file into Galaxy
- Go to your Galaxy instance of choice such as one of the usegalaxy.org, usegalaxy.eu, usegalaxy.org.au or any other. (This tutorial uses usegalaxy.org).
- Click Upload Data button:
- In the dialog box that would appear click “Choose local files” button:
- Find and select
SraRunInfo.csvfile from your computer- Click Start button
- Close dialog by pressing Close button
- You can now look at the content of this file by clicking galaxy-eye (eye) icon. You will see that this file contains a lot of information about individual SRA accessions. In this study every accession corresponds to an individual patient whose samples were sequenced.
Galaxy can process all 2,000+ datasets but to make this tutorial bearable we need to selected a smaller subset. In particular our previous experience with this data shows two interesting datasets SRR11954102 and SRR12733957. So, let’s pull them out.
Warning: Beware of CutsThe section below uses Cut tool. There are two cut tools in Galaxy due to historical reasons. This example uses tool with the full name Cut columns from a table (cut). However, the same logic applies to the other tool. It simply has a slightly different interface.
Hands-on: Creating a subset of data
- Find tool “Select lines that match an expression” tool in Filter and Sort section of the tool panel.
Galaxy may have an overwhelming amount of tools installed. To find a specific tool type the tool name in the tool panel search box to find the tool.
- Make sure the
SraRunInfo.csvdataset we just uploaded is listed in the param-file “Select lines from” field of the tool form.- In “the pattern” field enter the following expression →
SRR12733957|SRR11954102. These are two accession we want to find separated by the pipe symbol|. The|meansor: find lines containingSRR12733957orSRR11954102.- Click
Run Toolbutton.- This will generate a file containing two lines (well … one line is also used as the header, so it will appear the the file has three lines. It is OK.)
- Cut the first column from the file using tool “Advanced Cut” tool, which you will find in Text Manipulation section of the tool pane.
- Make sure the dataset produced by the previous step is selected in the “File to cut” field of the tool form.
- Change “Delimited by” to
Comma- In “List of fields” select
Column: 1.- Hit
ExecuteThis will produce a text file with just two lines:SRR12733957 SRR11954102
Now that we have identifiers of datasets we want and we need to download the actual sequencing data.
Download sequencing data with Faster Download and Extract Reads in FASTQ
Hands-on: Task description
- Faster Download and Extract Reads in FASTQ tool with the following parameters:
- “select input type”:
List of SRA accession, one per line
- The parameter param-file “sra accession list” should point the output of the tool “Cut” from the previous step.
- Click the
Run Toolbutton. This will run the tool, which retrieves the sequence read datasets for the runs that were listed in theSRAdataset. It may take some time. So this may be a good time to do get coffee.- Several entries are created in your history panel when you submit this job:
Pair-end data (fasterq-dump): Contains Paired-end datasets (if available)Single-end data (fasterq-dump)Contains Single-end datasets (if available)Other data (fasterq-dump)Contains Unpaired datasets (if available)fasterq-dump logContains Information about the tool execution
The first three items are actually collections of datasets. Collections in Galaxy are logical groupings of datasets that reflect the semantic relationships between them in the experiment / analysis. In this case the tool creates a separate collection each for paired-end reads, single reads, and other. See the Collections tutorials for more.
Explore the collections by first clicking on the collection name in the history panel. This takes you inside the collection and shows you the datasets in it. You can then navigate back to the outer level of your history.
Once fasterq finishes transferring data (all boxes are green / done), we are ready to analyze it.
Now what?
You can now analyze the retrieved data using any sequence analysis tools and workflows in Galaxy. SRA holds backing data for every imaginable type of *-seq experiment.
If you ran this tutorial, but retrieved datasets that you were interested in, then see the rest of the GTN library for ideas on how to analyze in Galaxy.
However, if you retrieved the datasets used in this tutorial’s examples above, then you are ready to run the SARS-CoV-2 variant analysis below.
Variation Analysis of SARS-Cov-2 sequencing data
In this part of the tutorial we will perform variant calling and basic analysis of the datasets downloaded above. We will start by downloading the Wuhan-Hu-1 SARS-CoV-2 reference sequence, then run adapter trimming, alignment and variant calling and finally look at the geographic distribution of some of the found variants.
Comment: The usegalaxy.* COVID-19 analysis projectThis tutorial uses a subset of the data and runs through the Variation Analysis section of covid19.galaxyproject.org. The data for covid19.galaxyproject.org is being updated continuously as new datasets are made public.
Get the reference genome data
The reference genome data for today is for SARS-CoV-2, “Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome”, having the accession ID of NC_045512.2.
This data is available from Zenodo.
Hands-on: Get the reference genome data
Import the following file into your history:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gz
- Copy the link location
Open the Galaxy Upload Manager (galaxy-upload on the top-right of the tool panel)
- Select Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
Adapter trimming with fastp
Removing sequencing adapters improves alignments and variant calling. fastp tool can automatically detect widely used sequencing adapters.
Hands-on: Task description
- fastp tool with the following parameters:
- “Single-end or paired reads”:
Paired Collection
- param-file “Select paired collection(s)”:
list_paired(output of Faster Download and Extract Reads in FASTQ tool)- In “Output Options”:
- “Output JSON report”:
Yes
Alignment with Map with BWA-MEM
BWA-MEM tool is a widely used sequence aligner for short-read sequencing datasets such as those we are analysing in this tutorial.
Hands-on: Align sequencing reads to reference genome
- Map with BWA-MEM tool with the following parameters:
- “Will you select a reference genome from your history or use a built-in index?”:
Use a genome from history and build index
- param-file “Use the following dataset as the reference sequence”:
output(Input dataset)- “Single or Paired-end reads”:
Paired Collection
- param-file “Select a paired collection”:
output_paired_coll(output of fastp tool)- “Set read groups information?”:
Do not set- “Select analysis mode”:
1.Simple Illumina mode
Remove duplicates with MarkDuplicates
MarkDuplicates tool removes duplicate sequences originating from library preparation artifacts and sequencing artifacts. It is important to remove these artefactual sequences to avoid artificial overrepresentation of single molecule.
Hands-on: Remove PCR duplicates
- MarkDuplicates tool with the following parameters:
- param-file “Select SAM/BAM dataset or dataset collection”:
bam_output(output of Map with BWA-MEM tool)- “If true do not write duplicates to the output file instead of writing them with appropriate flags set”:
Yes
Generate alignment statistics with Samtools stats
After the duplicate marking step above we can generate statistic about the alignment we have generated.
Hands-on: Generate alignment statistics
- Samtools stats tool with the following parameters:
- param-file “BAM file”:
outFile(output of MarkDuplicates tool)- “Set coverage distribution”:
No- “Output”:
One single summary file- “Filter by SAM flags”:
Do not filter- “Use a reference sequence”:
No- “Filter by regions”:
No
Realign reads with lofreq viterbi
Realign reads tool corrects misalignments around insertions and deletions. This is required in order to accurately detect variants.
Hands-on: Realign reads around indels
- Realign reads with lofreq tool with the following parameters:
- param-file “Reads to realign”:
outFile(output of MarkDuplicates tool)- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)- In “Advanced options”:
- “How to handle base qualities of 2?”:
Keep unchanged
Add indel qualities with lofreq Insert indel qualities
This step adds indel qualities into our alignment file. This is necessary in order to call variants using Call variants with lofreq tool
Hands-on: Add indel qualities
- Insert indel qualities with lofreq tool with the following parameters:
- param-file “Reads”:
realigned(output of Realign reads tool)- “Indel calculation approach”:
Dindel
- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)
Call Variants using lofreq Call variants
We are now ready to call variants.
Hands-on: Call variants
- Call variants with lofreq tool with the following parameters:
- param-file “Input reads in BAM format”:
output(output of Insert indel qualities tool)- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)- “Call variants across”:
Whole reference- “Types of variants to call”:
SNVs and indels- “Variant calling parameters”:
Configure settings
- In “Coverage”:
- “Minimal coverage”:
50- In “Base-calling quality”:
- “Minimum baseQ”:
30- “Minimum baseQ for alternate bases”:
30- In “Mapping quality”:
- “Minimum mapping quality”:
20- “Variant filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
The output of this step is a collection of VCF files that can be visualized in a genome browser.
Annotate variant effects with SnpEff eff:
We will now annotate the variants we called in the previous step with the effect they have on the SARS-CoV-2 genome.
Hands-on: Annotate variant effects
- SnpEff eff: tool with the following parameters:
- param-file “Sequence changes (SNPs, MNPs, InDels)”:
variants(output of Call variants tool)- “Output format”:
VCF (only if input is VCF)- “Create CSV report, useful for downstream analysis (-csvStats)”:
Yes- “Annotation options”: ``
- “Filter output”: ``
- “Filter out specific Effects”:
No
The output of this step is a VCF file with added variant effects.
Create table of variants using SnpSift Extract Fields
We will now select various effects from the VCF and create a tabular file that is easier to understand for humans.
Hands-on: Create table of variants
- SnpSift Extract Fields tool with the following parameters:
- param-file “Variant input file in VCF format”:
snpeff_output(output of SnpEff eff: tool)- “Fields to extract”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF[*].IMPACT EFF[*].FUNCLASS EFF[*].EFFECT EFF[*].GENE EFF[*].CODON- “multiple field separator”:
,- “empty field text”:
.
We can inspect the output files and see check if Variants in this file are also described in an observable notebook that shows the geographic distribution of SARS-CoV-2 variant sequences
Interesting variants include the C to T variant at position 14408 (14408C/T) in SRR11772204, 28144T/C in SRR11597145 and 25563G/T in SRR11667145.
Summarize data with MultiQC
We will now summarize our analysis with MultiQC, which generates a beautiful report for our data.
Hands-on: Summarize data
- MultiQC tool with the following parameters:
- In “Results”:
- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
fastp
- param-file “Output of fastp”:
report_json(output of fastp tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
Samtools
- In “Samtools output”:
- param-repeat “Insert Samtools output”
- “Type of Samtools output?”:
stats
- param-file “Samtools stats output”:
output(output of Samtools stats tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
Picard
- In “Picard output”:
- param-repeat “Insert Picard output”
- “Type of Picard output?”:
Markdups- param-file “Picard output”:
metrics_file(output of MarkDuplicates tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
SnpEff
- param-file “Output of SnpEff”:
csvFile(output of SnpEff eff: tool)
Conclusion
Congratulations, you now know how to import sequence data from the SRA and how to run an example analysis on these datasets.
Key points
Sequence data in the SRA can be directly imported into Galaxy
Frequently Asked Questions
Have questions about this tutorial? Check out the tutorial FAQ page or the FAQ page for the Variant Analysis topic to see if your question is listed there. If not, please ask your question on the GTN Gitter Channel or the Galaxy Help ForumUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Citing this Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials). http://0.0.0.0:4000/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html Online; accessed TODAY
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "", month = "", day = "" url = "\url{http://0.0.0.0:4000/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} Computational Biology} }
Galaxy Administrators: Install the missing toolsYou can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl http://localhost:4000//training-material//api/topics/variant-analysis/tutorials/sars-cov-2/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- {}